
Imagine being able to follow the journey of a virus as it moves, changes, and spreads through a community. This is the work of epidemiology—a field that provides crucial information that helps healthcare workers and governments respond effectively to pathogen outbreaks. Traditional approaches in epidemiology include methods like contact tracing, surveys, and statistical modelling. While these methods are effective, they can be time-consuming and labour-intensive, creating the risk of delays or overlooking important details.
But there is an alternative: analysing the genetic changes in virus samples collected from infected individuals. This allows scientists to construct evolutionary trees, similar to family trees, but for viruses. Specifically, scientists look at genetic information from virus samples to build an evolutionary tree that shows how a virus develops, spreads, and evolves over time. Just as a family tree helps trace ancestry, virus evolutionary trees help trace how pathogens like HIV, influenza, or COVID-19 evolve and move through populations.
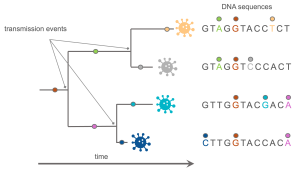
This scientific approach is known as phylodynamics; phylodynamics aims to reconstruct epidemiological processes from the shape of evolutionary (phylogenetic) trees. The core idea here is that the shapes of pathogen phylogenetic trees are affected by the disease dynamics; for instance, as the number of infections rises or falls, due to prophylactic measures such as vaccination, the shape of the tree changes accordingly. Phylodynamics uses the reverse approach: by analysing the shape of pathogen trees, it tries to uncover patterns in disease transmission.
What makes phylodynamics so powerful is its speed and precision. It allows researchers to quickly pinpoint outbreaks, identify superspreaders, and track the emergence of new viral variants. This means faster, more targeted responses to diseases, potentially saving countless lives and significantly improving public health outcomes.
Viral Phylodynamics and AI Approaches
Despite its many advantages, phylodynamics also has its limitations. Analysing pathogen evolutionary trees usually involves dealing with massive datasets—such as the GISAID COVID-19 dataset, which contains more than 16 million sequences (as of June 2025)—requiring extensive computational resources. That means complex and costly calculations, posing a significant challenge for researchers, especially those working with limited budgets. To address this issue, I introduced a new approach to phylodynamics, which uses AI to simplify and speed up this complex analysis. This new method, called PhyloCNN, is a neural network designed specifically to interpret evolutionary trees.
PhyloCNN uses a neural network method called a Convolutional Neural Network (CNN), which excels at detecting patterns within structured data, like images, audio, or, in this case, phylodynamics trees. The method directly encodes the tree topology (shape) by extracting information on the local context around each node in the tree (with a node representing the transmission events). Using this information, PhyloCNN can obtain key insights into how the disease spreads and evolves by estimating epidemiological parameters.
Unlike other AI tasks where ground truth data is available, such as image classification of, for instance, cats and dogs, where humans manually label the images used to train the neural network, training PhyloCNN required a different approach. We used simulated data, which allowed us to control all conditions of the disease evolution (including transmission rates, time to recovery, and the presence of superspreaders), which is not the case for trees obtained from real pathogens. We then trained a neural network to recognise phylodynamics trees that were generated under different disease dynamic conditions.
PhyloCNN in Action: HIV in Zurich
To demonstrate PhyloCNN’s effectiveness, I tested it on a real-world phylodynamics dataset from HIV infections in Zurich, focusing on superspreaders—individuals who transmit the virus at significantly higher rates. PhyloCNN results matched findings from extensive (and expensive) traditional studies. Even more impressively, PhyloCNN delivered these insights faster and at a lower computational cost. This efficiency means researchers can now tackle critical epidemiological questions faster and use fewer computational resources.
Importantly, since PhyloCNN is based on AI algorithms, it is also very flexible. It can be easily adapted to different viruses with specific characteristics; it can also incorporate more epidemiological information, such as contact tracing data or specific attributes from infected individuals that might influence disease spread.
Looking to the Future
PhyloCNN opens promising venues for epidemiological research. I am currently working with collaborators on extending this AI-driven approach to make it even more flexible and capable of analysing massive datasets (like the COVID-19 one mentioned above). The core advantages of AI-based phylodynamics lie in its flexibility and efficiency, allowing us to explore complex epidemiological models, including superspreading events, contact tracing, and quarantine strategies, which are often too computationally intensive for traditional phylodynamics methods. Our aim is to build on the foundations laid by PhyloCNN to create even faster and more adaptable tool for future disease outbreaks, enabling timely interventions.