Colon Cancer is not one disease — it is several different diseases that all look similar under the microscope, but got there by very different routes. Understanding these routes helps explain why some bowel cancers behave differently, respond to different treatments, and arise in different patients.
There are three main roads to bowel cancer.
Road 1 — The Classical Route (the most common)
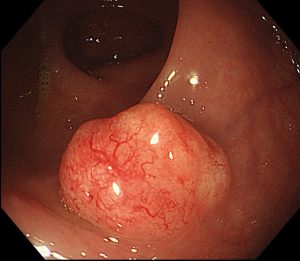
This is the familiar “polyp to cancer” story. A conventional polyp (adenoma) develops in the lining of the bowel and, over 10–15 years, accumulates a series of faults in key genes — first APC, then KRAS, then TP53. Each fault gives the cells a further growth advantage until cancer develops.
These cancers tend to be left-sided (descending colon and rectum) and are the ones most commonly detected by bowel cancer screening programmes.
Road 2 — The Serrated Route
This route starts from a different type of polyp — the sessile serrated lesion. These polyps are flat, pale, and easy to miss at colonoscopy, which is one reason this pathway is clinically important.
The first genetic fault here is in a gene called BRAF. What happens next is unusual — instead of accumulating more gene mutations, the tumour takes an epigenetic shortcut.
What does epigenetic mean?
Think of genes as light switches. A genetic mutation breaks the switch permanently. An epigenetic change is different — it puts a piece of “sticky tape” over the switch, keeping it off without actually breaking it. The gene is still physically intact but cannot be read. This chemical silencing process uses methyl groups and is called methylation.
In the serrated pathway, sticky tape is applied to the control switches of multiple genes simultaneously — a process called CIMP (CpG island methylator phenotype), or simply being “methylated”.
What happens next depends on which genes get silenced:
• If the sticky tape lands on MLH1 — the gene that proofreads and repairs DNA copying errors — the cell loses its ability to correct mistakes. Errors pile up rapidly in short repetitive DNA sequences called microsatellites. This is called microsatellite instability (MSI-H). These cancers have a very high number of mutations, attract a large immune response, tend to be right-sided, occur more often in older women, and respond very well to modern immunotherapy drugs.
• If the sticky tape does NOT silence MLH1, the cancer remains genomically stable (MSS). Paradoxically, this subgroup tends to behave more aggressively and does not respond to immunotherapy.
What about MSI-Low (MSI-L)?
You will sometimes see a third category — MSI-Low (MSI-L) — in reports and papers. This means there is a small degree of microsatellite instability, more than a fully stable tumour but nowhere near the level seen in MSI-H. In practice, MSI-L is a borderline or intermediate result. Most MSI-L tumours behave biologically more like MSS cancers than MSI-H cancers — they do not carry the same good prognosis, do not respond to immunotherapy, and are not generally associated with Lynch syndrome. MSI-L is thought to reflect minor, incidental errors in DNA copying rather than a true failure of the mismatch repair system. In most clinical algorithms, MSI-L is grouped with MSS for treatment decisions.
Road 3 — Lynch Syndrome (the inherited route)
Lynch syndrome also produces MSI-H cancers, but through a completely different mechanism. Here, a person is born with a faulty copy of one of the DNA repair genes. There is no sticky tape involved — the problem is hardwired into every cell of the body from conception.
How is it inherited?
Lynch syndrome follows an autosomal dominant pattern of inheritance. This means:
• Only one faulty copy of the gene is needed — inherited from one parent — to carry the syndrome. You do not need two faulty copies.
• Each child of an affected parent has a 50% chance of inheriting the faulty gene.
• It affects men and women equally.
• The faulty gene may have come from either the mother’s or father’s side of the family.
The genes involved are the mismatch repair genes: MLH1, MSH2, MSH6, and PMS2. Carrying a faulty copy does not guarantee cancer — it raises the lifetime risk substantially (up to 70–80% for bowel cancer with MLH1/MSH2 mutations) but is not inevitable. This is why Lynch families are offered regular surveillance colonoscopy.
Lynch syndrome also increases the risk of cancers in other organs — particularly the womb (endometrium), ovary, stomach, urinary tract, and small bowel — because the same repair machinery operates throughout the body.
The key distinction from the serrated pathway is that CIMP and BRAF mutation are absent in Lynch syndrome. Both arrive at MSI-H by different roads.
Detecting MMR Deficiency: IHC versus Molecular MSI Testing
In routine clinical practice, there are two completely different laboratory methods used to detect defective DNA repair in bowel cancers. They measure different things, and it is important not to confuse them.
Immunohistochemistry (IHC) — looking at the proteins
IHC is a staining technique performed by the pathologist on the tumour tissue section. Antibodies are applied that specifically bind to the four MMR proteins — MLH1, MSH2, MSH6, and PMS2 — and a colour reaction shows whether each protein is present or absent in the tumour cell nuclei.
In a normal tumour, all four proteins stain positively (present). If one or more proteins are lost, this indicates that the corresponding gene has been switched off or mutated — the cancer is said to show loss of MMR protein expression, or to be dMMR (deficient mismatch repair).
IHC has additional diagnostic value: the pattern of loss points towards the likely cause:
• Loss of MLH1 and PMS2 together → most likely sporadic (epigenetic silencing via CIMP, as in the serrated pathway). MLH1 methylation testing can confirm this.
• Loss of MSH2 and MSH6 together → strongly suggests Lynch syndrome (germline MSH2 mutation).
• Isolated loss of MSH6 or PMS2 → may indicate Lynch syndrome with a mutation in that specific gene.
• Loss of MLH1/PMS2 in a younger patient, or without BRAF mutation → raises suspicion for Lynch syndrome even if MLH1 is lost.
IHC is widely available, inexpensive, fast, and gives results the pathologist can interpret directly from the slide. However, it tests for protein — it tells you the protein is missing but does not directly measure what is happening to the DNA.
Molecular MSI Testing — looking at the DNA directly
Molecular MSI testing (also called PCR-based MSI testing or next-generation sequencing MSI analysis) works at the DNA level. It directly measures the lengths of specific short repetitive DNA sequences — microsatellites — in the tumour compared to normal tissue. If these sequences are abnormally variable in length, the tumour is called MSI-H. If they are stable, it is called MSS.
This test does not look at proteins at all. It confirms functionally that the DNA repair machinery has failed, regardless of which protein caused the problem or why.
So which test does what?
Think of it this way:
• IHC asks: which MMR protein is missing from the tumour? It identifies the defective component and points towards the mechanism (sporadic vs Lynch).
• Molecular MSI testing asks: has the loss of that protein actually caused DNA repair failure? It confirms the functional consequence.
• The two tests usually agree — a tumour that is dMMR by IHC is almost always MSI-H by molecular testing, and vice versa. But there are occasional discordant cases where one test is positive and the other negative, which is why both may be used in complex or equivocal situations.
• IHC is the standard first-line test in most pathology laboratories because it is practical and gives mechanistic clues. Molecular MSI testing is used to confirm, for clinical trials, or when IHC results are equivocal.
• Neither test on its own tells you whether the MMR deficiency is due to Lynch syndrome (germline, inherited) or sporadic methylation — that question requires germline genetic testing of the patient’s blood DNA, ideally guided by the IHC pattern and MLH1 methylation status.
The Bottom Line
• MSI (microsatellite instability) is the consequence — the end result of broken DNA repair, whatever the cause.
• MSI-H means the repair system has truly failed. MSI-L is a borderline finding that usually behaves like MSS and does not indicate Lynch syndrome.
• CIMP (methylation) is one mechanism that can cause MSI-H — by chemically silencing the MLH1 repair gene with sticky tape.
• The serrated pathway is the route — a distinct biological journey, starting from a different polyp, that frequently leads to CIMP and MSI-H.
• Lynch syndrome arrives at the same MSI-H destination by an entirely different, inherited road — a germline gene fault passed from parent to child with 50% probability.
• IHC and molecular MSI testing are complementary tools: IHC identifies which protein is lost and guides the search for the cause; molecular testing confirms that DNA repair has functionally failed.
Quick Reference Glossary
CIMP — CpG island methylator phenotype. The process of chemically silencing multiple genes simultaneously via methylation.
dMMR — Deficient mismatch repair. Detected by IHC as loss of one or more MMR proteins.
Epigenetic — Changes that affect gene activity without altering the DNA sequence itself.
IHC (Immunohistochemistry) — A staining technique that detects specific proteins in tissue sections using antibodies.
Methylation — Addition of methyl groups to gene promoters, acting like sticky tape over a switch to silence gene expression.
MMR — Mismatch repair. The cellular machinery that proofreads and corrects DNA copying errors.
MSI-H — Microsatellite instability-high. Confirms functional failure of DNA mismatch repair; high mutation burden; responds to immunotherapy.
MSI-L — Microsatellite instability-low. A borderline finding; behaves clinically like MSS; not associated with Lynch syndrome.
MSS — Microsatellite stable. Normal DNA repair function.
Autosomal dominant — Inheritance pattern where one faulty copy of a gene (from either parent) is sufficient to cause the condition; 50% transmission risk per child.
Lynch syndrome — An inherited condition caused by a germline mutation in an MMR gene (MLH1, MSH2, MSH6, or PMS2), predisposing to bowel and other cancers.
BRAF / KRAS / APC / TP53 — Genes that when mutated drive cancer development along different pathways.
Reference: Guinney et al., Nature Medicine 2015 (Consensus Molecular Subtypes) | WHO Classification of Digestive System Tumours
Read A plain-language guide to the molecular pathways in colorectal cancer in full